近日,大连化物所催化基础国家重点实验室理论催化创新特区研究组(05T8组)肖建平研究员团队与浙江大学肖丰收教授和王亮研究员团队合作,在合成气一步法制备乙醇的研究中取得新进展,开发了一套新的理论方法来理解合成气选择性制备乙醇的反应路径和分析产物选择性。
乙醇是重要的化学品和燃料。通过合成气高效并高选择性地直接制备乙醇是目前能源研究领域的热点。催化剂反应性过强或过弱都会造成副产物选择性过高,因此合成气直接制备乙醇也是难点。深入理解合成气转化的反应机理,对后续提升催化剂的活性和寻找非贵金属催化剂具有重要意义。合成气转化过程中有数十种中间体及过渡态,数千种反应路径,而传统的理论方法不能很好地理解这一过程。基于此,肖建平团队开发了一套反应路径研究和产物选择性分析的新方法,可以将数十种中间体以及过渡态的吸附能简化到两个维度——CO和OH的吸附能,并以此来描述不同催化剂对产物的选择性(如下图的二维反应相图),进而理解和指导催化剂的设计。此方法还能综合热力学和动力学影响自动搜寻生成乙醇的最优反应路径。
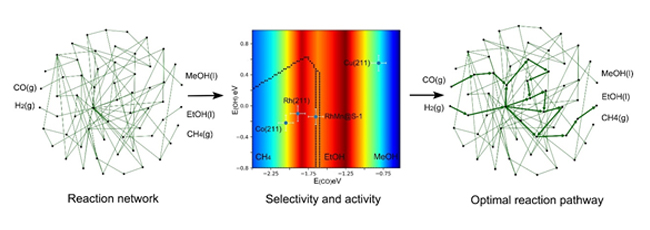
在合成气转化过程中,如果CO分子和催化剂(如纯Rh、Co催化剂等)结合太强,碳-氧键基本全部打断,进而选择性得到甲烷;如果CO和催化剂(如Cu催化剂)结合太弱,碳-氧键完全不能被打断,所以只能得到甲醇。CO和催化剂之间的结合能力只有在非常小的一个能量窗口时,CO解离之后的中间体(CHx*)和未解离的CO*/CHO*共存,才能有较好的乙醇选择性。理论计算结果表明,CH2*和CO/CHO*中间体的耦合是最关键的步骤,MnOx结构和分子筛提供的限域环境影响了界面处(MnOx附近)的Rh原子的电子结构,使RhMn@S-1催化剂恰巧落在这个区间,最终使CH2*和CO*/CHO*中间物质能大量共存,从而得到较高的乙醇选择性。该工作为设计高选择性合成气制备乙醇的催化剂提供了新思路。
相关成果发表在Chem上。该工作得到了国家重大研发计划、国家自然科学基金、中科院重点部署项目和辽宁省“兴辽英才计划”等研究经费的支持。

