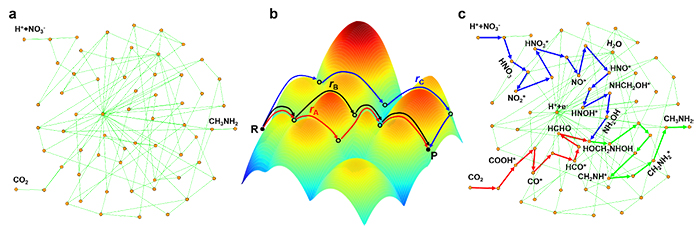
Carbon dioxide (CO2) and nitrate (NO3–) co-reduction to valuable chemicals, e.g., methylamine (MMA) production, by electricity generated from renewable energy sources, has been considered as a promising route for electrochemical synthesis. However, the mechanism of C–N coupling in the process is key and remains unclear. Herein, we studied the (quasi) activity trend over a set of metal phthalocyanine (MPc) via density functional theory (DFT) calculations and reaction phase diagram analysis. Surprisingly, we found that the lowest barrier for C–N coupling is via desorbed HCHO and NH2OH rather than either being adsorbed on the catalyst. Then, we performed kinetic barrier calculations and microkinetic modeling over CoPc–NH2, which was previously observed with the capability of producing MMA, to understand the Faradaic efficiency trends of MMA and other products at different potentials. Our kinetic model provides important insights for improving the MMA activity and selectivity and is useful for catalyst design for C–N coupling in general.