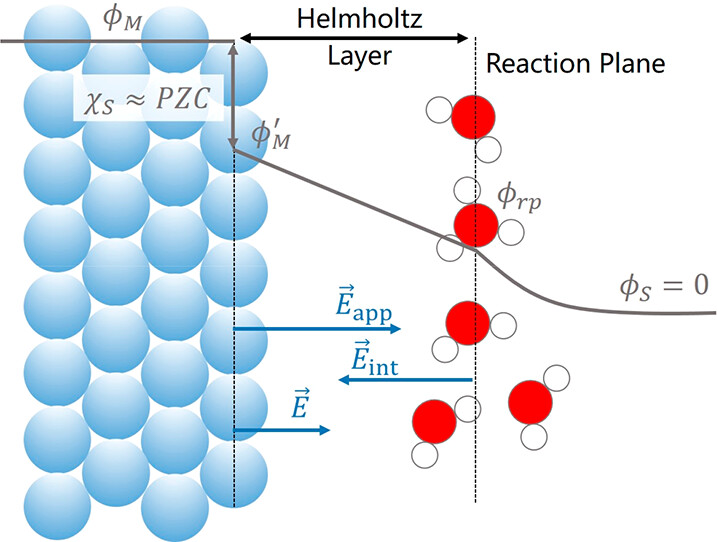
Electrochemical interfaces are grand canonical ensembles of varying electrons. Simulating them by standard first-principles methods is a challenging task, since the number of electrons (or charge) is fixed in the calculation. Under the constant charge framework, we developed a constant potential simulation method realized by adding an adaptive electric field to a charge neutral system. Electric field is the controlling variable. In addition, we defined an internal reversible hydrogen electrode potential (ϕIRHE), which can ensure the model independence of our method. To validate our method, the reaction energies of some electrochemical reactions are calculated, the results are comparable with the computational hydrogen electrode model and experiments. At last, the evolution of transition state structures and charge transfer coefficients of some electrochemical reactions on Ag(111) surface were discussed by our method.